
|
Photophysics and Photochemistry of Transition Metal Compounds |
| Home Research Members Collaborations Publications |



|
 |
|||||||
This work illustrates a simple approach for optimizing long-lived near-infrared lanthanide-centered luminescence using trivalent chromium chromophores as sensitizers. Reactions of the segmental ligand L2 with stoichiometric amounts of M(CF3SO3)2 (M = Cr, Zn) and Ln(CF3SO3)3 (Ln = Nd, Er, Yb) under aerobic conditions quantitatively yield the D3-symmetrical trinuclear [MLnM(L2)3](CF3SO3)n complexes (M = Zn, n = 7; M = Cr, n = 9), in which the central lanthanide activator is sandwiched between the two transition metal cations. Visible or NIR irradiation of the peripheral Cr(III) chromophores in [CrLnCr(L2)3]9+ induces rate-limiting intramolecular intermetallic Cr→Ln energy transfer processes (Ln = Nd, Er, Yb), which eventually produces lanthanide-centered near-infrared (NIR) or IR emission with apparent lifetimes within the millisecond range. As compared to the parent dinuclear complexes [CrLn(L1)3]6+, the connection of a second strong-field [CrN6] sensitizer in [CrLnCr(L2)3]9+ significantly enhances the emission intensity without perturbing the kinetic regime. This work opens novel exciting photophysical perspectives via the buildup of non-negligible population densities for the long-lived doubly excited state [Cr*LnCr*(L2)3]9+ under reasonable pumping powers. | ||||||||
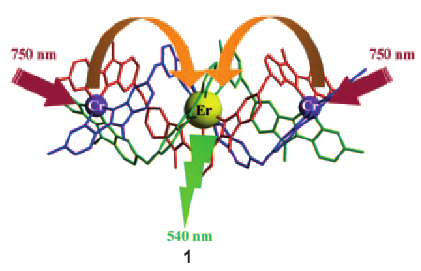 |
|
|||||||
The connection of two CrIII sensitizers around a central ErIII acceptor in a self-assembled cation provides high local metal concentrations that favor efficient nonlinear energy transfer upconversion luminescence (see picture). Upon selective low-energy near-infrared irradiation of CrIII-centered transitions, 1 displays an unprecedented molecular two-photon upconverted green ErIII-centered emission. | ||||||||

|
 |
|||||||
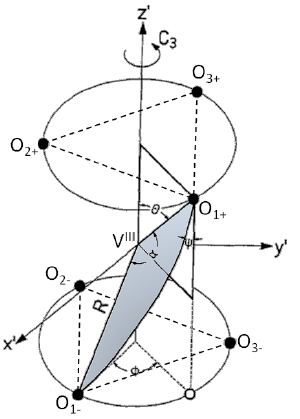
The electronic structure and the photophysical properties of the vanadium(III)ion in pseudo-octahedral oxygen coordination is reviewed. V3+ has received much interest from spectroscopists in recent years due to the advancement of state-of-the-art experimental techniques such as inelastic neutron scattering and high-field electron paramagnetic resonance spectroscopy that directly interrogate its large ground state zero-field splittings (ZFSs) and to rational parameterization of the ligand fieldp arameters using the angular overlap model. However, for V3+ these ZFSs can be large enough to also be probed directly by high-resolution electronic absorption spectroscopy of intra-configurational (t22g → t22g) spin-forbidden transitions in the near-IR and visible regions. The luminescent properties of V3+ with hexa-oxo and tris-bidentate di-oxo-coordination are quite disappointing compared to its neighbor in the periodic table, Cr3+, in similar environments. The efficient non-radiative pathways in these compounds are reviewed and compared to recent work on V3+ doped into NaMgAl(ox)3⋅9H2O. The poor luminescence quantum efficiencies of V3+ oxo complexes is explained by strong coupling of multi-phonon processes with a dynamic Jahn-Teller distortion originating from the 3E trigonal component of the 3T1g ground state. | ||||||||
 |
|
|||||||
The ground-state electronic structures of K3V(ox)3·3H2O, Na3V(ox)3·5H2O, and NaMgAl1–xVx(ox)3·9H2O (0 < x <= 1, ox = C2O42–) have been studied by Fourier–transform electronic absorption and inelastic neutron scattering spectroscopies. High-resolution absorption spectra of the 3Γ(t2g2) → 1Γ(t2g2) spin-forbidden electronic origins and inelastic neutron scattering measurements of the pseudo-octahedral [V(ox)3]3– complex anion below 30 K exhibit both axial and rhombic components to the zero-field-splittings (ZFSs). Analysis of the ground-state ZFS using the conventional S = 1 spin Hamiltonian reveals that the axial ZFS component changes sign from positive values for K3V(ox)3·3H2O (D ≈ +5.3 cm–1) and Na3V(ox)3·5H2O (D ≈ +7.2 cm–1) to negative values for NaMgAl1–xVx(ox)3·9H2O (D ≈ –9.8 cm–1 for x = 0.013, and D ≈ –12.7 cm–1 for x = 1) with an additional rhombic component, |E|, that varies between 0.8 and 2 cm–1. On the basis of existing crystallographic data, this phenomenon can be identified as due to variations in the axial and rhombic ligand fields resulting from outer-sphere H-bonding between crystalline water molecules and the oxalate ligands. Spectroscopic evidence of a crystallographic phase change is also observed for K3V(ox)3·3Y2O (Y = H or D) with three distinct lattice sites below 30 K, each with a unique ground-state electronic structure. | ||||||||
|
 |
|||||||
High-resolution Fourier transform absorption and luminescence spectroscopy reveal axial and rhombic zero-field splittings of the spin-forbidden electronic origins of V3+ in NaMgAl(ox)3·9H2O (ox=oxalate) single crystals below 25 K. The temperature dependence of the integrated absorption of the split features display behavior consistent with a Boltzmann distribution within the zero-field split 3Â2 ground state of V3+. Weak luminescence is observed in the near-IR from the lowest energy spin-forbidden transition with a luminescence lifetime of less than 0.5 μs at 11 K and an estimated quantum efficiency of the order of 10-5 | ||||||||
Download this list in format RIS
 EndNote
EndNote  BibTex
BibTex  PDF XML
PDF XML Last update Friday December 08 2017